Clonal lineage trees (affinity maturation)¶
Where expand_clones produces a star - one founder and many independent descendants -
clonal_lineage grows a real tree: a generation-by-generation
birth–death process in which cells divide, somatically hypermutate, are selected
for antigen affinity, and are finally sampled. The output is a set of
per-cell AIRR records plus the ground-truth lineage tree (topology,
ancestral sequences, abundances) - the kind of object B-cell lineage-inference tools
(GCtree, IgPhyML, dowser, Change-O) are built to reconstruct. This page explains
precisely how it works under the hood; nothing here is a black box.
Why a tree, not a star¶
A clonal family in vivo is the progeny of one naive B cell that has entered a germinal center. Inside that germinal center the cell divides, its B-cell receptor somatically hypermutates a few bases per division, and cells whose mutated receptor binds the antigen better are preferentially expanded (affinity maturation). The result is a genealogy with internal ancestors, unequal branch lengths, and selective sweeps - a tree.
The older expand_clones collapses all of that into a star: it takes the founder
recombination and draws per_clone independent descendants directly off it. That
is fine for "many reads that share a V(D)J truth", but it has no genealogy, no
generations, no selection, and no ancestral nodes - so it cannot serve as ground
truth for lineage reconstruction. clonal_lineage adds the missing biology.
clonal_lineageis BCR-only. T cells do not somatically hypermutate, andclonal_lineageapplies S5F SHM, so calling it on a TCR locus raises a clearValueError. A TCR "clone" is one rearrangement proliferated to many identical copies; the meaningful quantity is the clone-size distribution, not a mutation tree. For TCR and flat clonal repertoires, useclonal_repertoire- it draws a heavy-tailed clone size per clone and emitsclone_id+duplicate_count(see Clone-size distributions).
Quick start¶
import GenAIRR as ga
result = (
ga.Experiment.on("human_igh")
.recombine() # the founder: one V(D)J recombination per clone
.clonal_lineage(
n_clones=20, # grow 20 independent families
max_generations=6, # germinal-center rounds
n_max=300, # per-generation living-population carrying capacity
n_sample=30, # cells sampled per family at the end
rate=0.01, # per-base S5F SHM rate, per division
lambda_base=1.6, # mean offspring per cell per generation
selection_strength=10.0, # 0 = neutral; >0 = affinity selection
)
.run_records(seed=0)
)
# Per-cell AIRR records, tagged with clone + lineage metadata:
for rec in result.records[:3]:
print(rec["clone_id"], rec["lineage_node_id"], rec["lineage_generation"],
rec["lineage_abundance"], rec["lineage_affinity"], rec["v_call"])
# Ground-truth trees, one per clone:
tree = result.lineage_trees[0]
tree.validate() # structural invariants (raises if malformed)
newick = tree.to_newick() # true topology, branch length = per-edge mutations
fasta = tree.to_fasta() # every node's sequence (ancestral + observed)
table = tree.to_node_table_tsv()
How it works under the hood¶
Each clone is grown independently by a generation-synchronous birth–death process
in the Rust engine. The whole loop is deterministic for a given seed.
flowchart TB
A["Founder: one V(D)J recombination<br/>(the pre-fork plan, run once per clone)"] --> B["Generation g = 1..max_generations"]
B --> C["For each live cell:<br/>offspring k ~ Poisson(λ_eff)"]
C --> D["λ_eff = lambda_base<br/>× carrying-capacity damping<br/>× affinity fitness(cell)"]
C --> E["Each child = parent + per-division S5F mutations"]
E --> F["Child affinity = exp(−β · weighted aa distance to target)"]
F --> B
B --> G["Stop at max_generations / extinction / capacity"]
G --> H["Sample n_sample cells from the final population"]
H --> I["Collapse identical genotypes → abundances<br/>(observed nodes)"]
I --> J["Project each observed cell → AIRR record<br/>+ emit ground-truth tree"]1. The founder¶
recombine() (everything before clonal_lineage in the chain) is the per-clone
phase: it runs once to produce the naive rearrangement - the V/D/J allele picks,
trims, NP bases, junction. That founder Simulation (and its recombination trace)
is the root of the tree. Clone c uses seed seed + c × 1_000_000, so families
are independent and reproducible.
2. Generation-synchronous birth–death¶
Growth proceeds in discrete generations. In each generation every currently-live cell produces a number of offspring drawn from a Poisson distribution:
k = 0 means the cell leaves no progeny (it becomes a tip); k ≥ 1 creates k
children for the next generation. λ_eff is the base offspring rate lambda_base
modulated by two factors below.
3. Carrying capacity (logistic damping)¶
A germinal center is population-bounded, so the effective rate is damped as the
live population P approaches n_max:
Near saturation λ_eff → 0 and growth plateaus instead of exploding. A hard cap
also prevents the live set from exceeding n_max even on a lucky Poisson draw.
4. Per-division somatic hypermutation (S5F)¶
Every child is a clone of its parent's Simulation with a fresh round of somatic
hypermutation applied. GenAIRR reuses its context-sensitive S5F engine (the
same one behind mutate(model="s5f")): mutations are drawn from the 5-mer
mutability/substitution kernel (s5f_model, default "hh_s5f") at per-base rate
rate, applied one at a time with the sequence context re-evaluated between
mutations. Because SHM only substitutes bases in place, each cell keeps the
founder's V/D/J assignments and region map - its germline ancestry stays intact,
which is what lets every node be projected to a correct AIRR record (below).
The branch length stored on each edge is the realized number of substitutions introduced on that division.
5. Affinity selection¶
This is what turns a neutral tree into affinity maturation. Each cell has an affinity to a target sequence:
What "affinity" means here - read this. This is a sequence-distance proxy, not a physically modeled antigen-binding affinity. It is a BLOSUM62 substitution-aware amino-acid distance from the cell's translated receptor to a target amino-acid sequence, mapped through
exp(−β · distance). There is no Kd, no antigen concentration, no biophysical binding model anywhere in the computation. Treat it as a tunable selection pressure that pulls the lineage toward a target sequence - the closer a cell's receptor gets to the target, the higher its "affinity" and the faster it divides.
weighted_aa_distance is a BLOSUM62 substitution-aware amino-acid distance
between the cell's translated receptor and the target (region weights let CDRs be
emphasized; v1 uses uniform weights, with CDR3-weighting as a planned refinement).
affinity is 1.0 at the target sequence and decays toward 0 as the cell diverges.
The target is either supplied by you (target_aa=..., a target amino-acid
sequence) or auto-generated as a "mature" target - the founder's amino-acid
sequence with mature_substitutions random residue changes (the standard
benchmark convention).
Affinity feeds back into the offspring rate through a fitness multiplier:
fitness = max(0, 1 + selection_strength · (affinity − founder_affinity))
λ_eff = lambda_base × carrying_capacity_damping × fitness
founder_affinity is the affinity of the naive founder, so the founder has
fitness ≈ 1, cells that improve on it divide faster, and worse cells divide
slower - producing selective sweeps. selection_strength = 0 makes fitness ≡ 1,
i.e. a neutral tree (byte-identical to growing with no selection at all).
Calibration note.
exp(−beta · distance)can underflow to ~0 for long receptor sequences at largebeta, flattening selection. If you supply a full antigentarget_aa, keepbetasmall (e.g.1e-3) or use the auto target.
6. Sampling and genotype collapse¶
When growth stops (at max_generations or capacity), n_sample cells are sampled
from the LIVING final-generation population - the cells that are alive when
growth stops. Cells with identical genotypes are then collapsed into
observed cells: the first cell seen for a genotype becomes the observed
representative and accumulates an abundance count (surfaced as both
lineage_abundance and the AIRR-standard duplicate_count) - so abundance-aware
tools (GCtree, Change-O, SCOPer, dowser) get observed cells with multiplicities.
The observed cells are the ones that become AIRR records.
Because sampling draws from the living population, an extinct clone - one
whose founder draws 0 offspring - has no living cells and therefore yields zero
observed cells and zero records. A single founder at lambda_base ≈ 1.5 goes
extinct roughly 25 % of the time. By default (allow_extinction=False) each
requested clone is conditioned on survival: an extinct family is re-grown with
a fresh deterministic sub-seed (up to a bounded number of attempts) so you reliably
get all n_clones families back. Determinism is preserved - the same top-level
seed always reproduces the same result. Set allow_extinction=True to accept
extinction instead: extinct clones are skipped and you get fewer than n_clones
families.
The full genealogy - including every unobserved internal ancestor - is still
emitted in the LineageTree (and its Newick/FASTA), so ancestral-sequence
reconstruction can be scored against truth; note, however, that observed/sampled
nodes are always tips, not internal ancestors (direct sampling of internal
ancestors is a future addition).
What you get back¶
Per-cell AIRR records¶
result.records is a list of full AIRR Rearrangement dicts, one per observed
(genotype-collapsed) cell. Each carries the founder's recombination provenance
(v_call, d_call, j_call, junction, …) - correct because the node's Outcome
reuses the founder's recombination trace - plus its own mutated sequence. Mutation
counts (n_mutations, n_v_mutations, …, and the IMGT-subregion counters) are
recomputed from the cell's sequence vs. germline - these are net differences
from germline (accumulated across all divisions from founder to leaf). Because
identical genotypes are collapsed before sampling, the number of records per clone
is ≤ n_sample; the lineage_abundance field (mirrored by the AIRR-standard
duplicate_count) accounts for the collapsed copies.
Branch lengths vs. record
n_mutations. Newick branch lengths (as returned byto_newick()) count the per-division substitution events along each edge - re-mutations of a site that was already mutated count again. The record fieldn_mutationsis the net difference from germline at the leaf (a site mutated and then back-mutated is not counted). As a result, summing branch lengths root→leaf will generally exceed a leaf'sn_mutations. Both quantities are standard and correct: branch lengths track evolutionary distance along an edge (as tools like GCtree and IgPhyML expect), whilen_mutationstracks the observable deviation from germline (as AIRR requires).
Consistency check: n_v + n_d + n_j + n_np == n_mutations holds for every record.
Lineage metadata stamped on every record:
| Field | Meaning |
|---|---|
clone_id |
Which family (0 … n_clones−1) - the ground-truth clone label |
lineage_node_id |
The cell's node id within its tree |
lineage_parent_id |
Parent node id (−1 for the founder) |
lineage_generation |
Generation depth (founder = 0) |
lineage_abundance |
Observation count after genotype collapse |
duplicate_count |
AIRR-standard alias of lineage_abundance (read by Change-O / SCOPer / dowser) |
lineage_affinity |
Sequence-distance proxy to the target (see §5). 0 only when no target is in play - fully neutral mode (target_aa=None and selection_strength=0). If a target_aa is supplied (or selection is on), affinities are computed and reported even when selection_strength=0 |
Ground-truth lineage trees¶
result.lineage_trees is one LineageTree per clone. Each tree exposes the full
genealogy (every node, not just observed ones) and three exporters consumed by
inference tools:
to_newick()- standard rooted Newick; the founder is the root, branch lengths are per-edge mutation counts. (((node3:1)node1:1,node2:1)node0;)to_fasta()- every node's sequence, ancestral and observed, so ancestral-sequence-reconstruction can be scored against truth.to_node_table_tsv()-node_id, parent_id, generation, mutations_from_parent, abundance, observed, affinity, sequence.nodes()/validate()- node access and a structural-invariant check.
Library-prep & sequencing artefacts¶
Library-prep and sequencing passes can follow clonal_lineage - they run
independently on each observed cell, so every read picks up its own noise:
result = (ga.Experiment.on("human_igh").recombine()
.clonal_lineage(n_clones=10, n_sample=30, rate=0.01, selection_strength=10)
.sequencing_errors(rate=0.005)
.pcr_amplify(rate=0.002)
.run_records(seed=0))
Each observed cell's post-SHM sequence is passed through the corruption plan with
its own seed, and the resulting artefacts are merged back onto the cell's record.
The founder's recombination provenance (v_call, d_call, j_call, trims,
junction) and the per-segment SHM counts are preserved; the record additionally
reports the artefact counters (n_quality_errors, n_pcr_errors, n_indels, …).
Supported passes are the per-read library-prep / sequencing artefact set also
used by clonal_repertoire and legacy expand_clones:
sequencing_errors, pcr_amplify, polymerase_indels, end_loss_*,
ambiguous_base_calls, random_strand_orientation.
mutate is not allowed after clonal_lineage - SHM is internal to the lineage
engine (set it via clonal_lineage(rate=...)). paired_end is not allowed yet
either (the read layout is not wired through the per-cell corruption merge - a future
addition).
Validation works on lineage results too: run_records(..., validate_records=True)
runs the per-record postcondition check and the clonal-family consistency check
(by clone_id), with or without a corruption pass. run_records(...,
expose_provenance=True) adds truth_v_call / truth_d_call / truth_j_call
columns from the founder assignments, and result.outcomes carries the per-record
Outcome objects index-aligned with result.records.
Clone-size distributions (TCR and repertoire mix)¶
For TCR, use
clonal_repertoire.clonal_lineageitself is BCR-only - it still rejects TCR loci. The heavy-tailed clone-size model described below is now exposed as a fluent DSL workflow viaclonal_repertoire; that is the TCR (and flat-BCR-abundance) path. This section explains the model; the dedicated guide is the place to drive it.
Real repertoires are not uniform: a few clones are huge, most are singletons.
clonal_repertoire draws clone sizes from a
heavy-tailed distribution (rounded power-law / Zipf-like by default, log-normal
optional) with a controllable unexpanded fraction (size-1, never-expanded
clones). For TCR -
which has no SHM - a clone is simply one rearrangement at copy-number size, with
within-clone variation coming only from the post-fork sequencing/PCR-error passes;
identical reads collapse into AIRR records carrying clone_id + duplicate_count.
That mixes large expanded families with a realistic singleton tail. See the
Clonal repertoires guide for the full workflow.
Determinism¶
Everything is keyed on seed. Clone c grows from seed + c × 1_000_000;
within a clone, generations, divisions, mutations, and sampling all draw from
seeded RNG streams (growth and sampling use separate streams so they don't
interfere). Re-running with the same seed reproduces the trees and records
byte-for-byte.
Parameters¶
| Parameter | Default | Meaning |
|---|---|---|
n_clones |
- | Number of independent families to grow |
max_generations |
10 | Germinal-center rounds (≤ 1000) |
n_max |
1000 | Per-generation LIVING-population carrying capacity - the live population each generation is capped at this. It is not a hard cap on total cells per clone; the tree can contain more total nodes across generations |
n_sample |
50 | Cells sampled per family at the end; records per clone ≤ this (genotype-collapsed) |
rate |
0.05 | Per-base S5F SHM rate, per division |
lambda_base |
1.5 | Mean offspring per cell per generation |
selection_strength |
0.0 | 0 = neutral drift (fitness ≡ 1); set > 0 for affinity selection. Note 0 makes selection neutral but does not force lineage_affinity to 0 - affinities are still computed/reported whenever a target_aa is supplied |
beta |
1.0 | Affinity steepness in exp(−beta·distance) |
target_aa |
None |
Target amino-acid sequence (a full translated receptor) used as the selection target (BLOSUM62-weighted distance, position-wise; only the overlapping prefix is scored when lengths differ). A sequence-distance proxy, not a biophysical antigen. None ⇒ auto "mature" target |
mature_substitutions |
5 | aa substitutions for the auto target |
s5f_model |
"hh_s5f" |
Bundled S5F kernel |
allow_extinction |
False |
False ⇒ condition each clone on survival (retry extinct founders with fresh deterministic sub-seeds), so you reliably get n_clones families. True ⇒ accept extinction and skip extinct clones, producing fewer families |
Clone recovery: what we actually ran¶
The point of planting ground-truth clones is that other people's tools can find them. Two clusterers were actually run against the planted labels and both recover them perfectly (adjusted Rand index = 1.0) at realistic SHM:
- Immcantation Change-O
DefineClonesat its default junction-distance threshold (0.16) recovers the planted clones exactly. - An in-repo, implementation-independent standard-heuristic clusterer (V/J + junction-length + single-linkage) recovers them just as cleanly.
The export formats are designed to feed the broader B-cell lineage ecosystem -
tree-based tools like GCtree, IgPhyML, and dowser consume the
Newick/FASTA/node-table exports, and abundance-aware clustering tools like
SCOPer read the AIRR TSV with duplicate_count. Those tools were not run
as part of this validation; the claim here is scoped to the two clusterers above
and to format compatibility, not to having executed the full ecosystem.
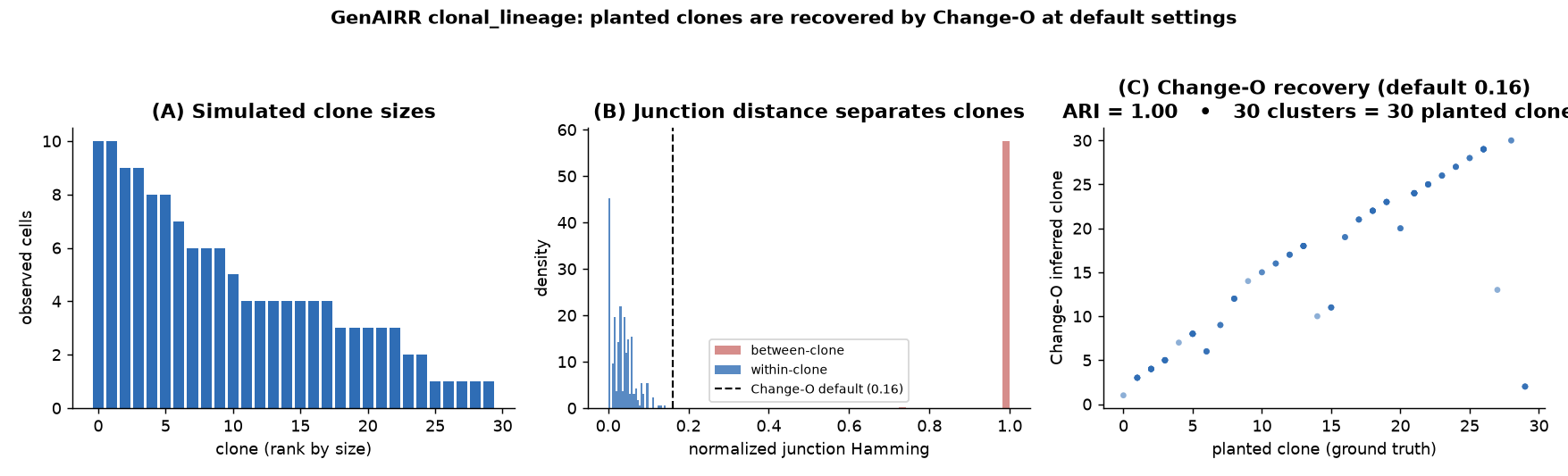
30 clones simulated with clonal_lineage (human IGH, realistic SHM). (A) the
repertoire has a realistic spread of clone sizes. (B) within-clone junction
distances sit far below Change-O's default 0.16 threshold while between-clone
distances are ~1.0 - the planted clones are cleanly separable. (C) Change-O at
its default threshold recovers all 30 planted clones (adjusted Rand index = 1.0,
precision = recall = 1.0).
Reproduce it: run Change-O on a simulated repertoire¶
import GenAIRR as ga
import pandas as pd
result = (ga.Experiment.on("human_igh").recombine()
.clonal_lineage(n_clones=30, max_generations=3, n_max=300,
n_sample=20, rate=0.008, lambda_base=1.6,
selection_strength=0.3)
.run_records(seed=42))
# Write an AIRR-format TSV. Keep the ground-truth clone label under a NON-AIRR
# name so it doesn't collide with the tool's inferred `clone_id` column.
df = pd.DataFrame(result.records)
df = df.rename(columns={"clone_id": "true_clone_id"})
df.to_csv("repertoire.tsv", sep="\t", index=False)
# Immcantation Change-O (pip install changeo) infers clones from junctions:
DefineClones.py -d repertoire.tsv --format airr \
--act set --model ham --norm len --dist 0.16 -o clones.tsv
Comparing Change-O's inferred clone_id against the planted true_clone_id
(e.g. with sklearn.metrics.adjusted_rand_score) gives ARI = 1.0 - a perfect
match.
What the validation shows across SHM regimes¶
- Perfect precision, always. Across every tool and threshold tested, two different planted clones are never merged. The per-clone founding rearrangements are distinct, so the planted signal is unambiguous.
- Full recovery at a matched threshold. Detection is a function of how mutated
the lineage is versus the caller's distance cutoff. At realistic SHM, the default
0.16 cutoff recovers everything; for deeply matured lineages (e.g.
rate=0.05, 6 generations → ~21 % SHM) you raise the cutoff (a threshold sweep climbs from ARI 0.26 at 0.16 → 0.91 at 0.30 → 1.0 at 0.45). This mirrors how these tools behave on real data and is not a property of the simulator. - Independent of any one tool. The same recovery holds for the in-repo
implementation-independent V/J + junction-length + single-linkage clusterer - so
the signal is not an artefact of Change-O's specific model. The exported
Newick/FASTA/AIRR-TSV (with
duplicate_count) are designed to feed tree-based and abundance-aware methods (GCtree, IgPhyML, dowser, SCOPer) directly; those downstream tools were not run here.
In short: the two clusterers we ran recover the planted clones exactly, and the export formats are built to hand the same ground truth to the wider ecosystem.
Relationship to expand_clones¶
expand_clones (the star model) is deprecated but still works - it remains
useful for "many reads sharing one V(D)J truth" without a genealogy.
clonal_lineage is not a drop-in replacement. It grows real
affinity-maturation trees rather than a flat star, so the surface differs:
- Different parameters. There is no
per_clone; the number of observed records depends onn_sample, genotype collapse, and selection (not a fixedn_clones × per_cloneproduct). SHM is internal (rate=...), not a separatemutatestep. - Different return shape.
clonal_lineagereturns aSimulationResultWithLineageswith per-clone.lineage_trees(Newick / FASTA / node-table exporters) alongside the per-cell records.
What does carry over: the same per-read library-prep / sequencing passes
(sequencing_errors, pcr_amplify, …) can follow clonal_lineage exactly as they
follow other clonal workflows, applied independently per observed cell (see
Library-prep & sequencing artefacts). And
run_records(..., validate_records=True) is supported on lineage results too.