Benchmarking genotype inference¶
The point of simulating from a known genotype is that you can run a genotype-inference tool on the resulting repertoire and score it against the planted truth - with no real-data uncertainty about what the right answer is.
The recipe is the same for any tool:
- Build a
Genotype, simulate a repertoire, write the AIRR table (result.to_tsv(...)) and/or reads FASTA, and the ground truth (genotype.to_tsv(...)). - Run the inference tool to recover the per-individual allele set.
- Compare recovered vs planted: presence/absence, zygosity, and (for discovery tools) any novel alleles.
Worked example: TIgGER and IgDiscover recover a planted genotype¶
To show this end to end we planted a diploid IGH genotype in human_igh -
3 heterozygous V genes (two alleles each), 3 homozygous (one allele),
and 3 fully deleted genes - and filled the rest from the reference. We
simulated 4,000 reads with light SHM, then ran two independent AIRR
genotype-inference tools on the result:
TIgGER (Immcantation; consumes the AIRR
table) and IgDiscover (germline discovery from the
raw reads, with its own IgBLAST).
Because GenAIRR already emits AIRR records with v_call and
sequence_alignment, TIgGER's inferGenotype consumes the rearrangement table
directly - no separate IgBLAST step is needed:
library(tigger); library(airr)
rep <- read_rearrangement("repertoire.tsv") # GenAIRR's AIRR output
germ_v <- readIgFasta("germline_V.fasta") # cartridge V germline (names match v_call)
# find_unmutated=TRUE asks inferGenotype to base calls on unmutated reads per
# allele; the light-SHM simulation below provides them.
geno <- inferGenotype(rep, germline_db = germ_v, find_unmutated = TRUE)
plotGenotype(geno)
The germline_V.fasta written below is ungapped - fine for inferGenotype;
if you go on to TIgGER's IMGT-gapped steps (reassignAlleles) supply a gapped V
germline instead.
TIgGER recovered the planted genotype exactly: every heterozygous gene → two alleles, every homozygous gene → one, every deleted gene → absent. Across all 52 V genes, allele-presence precision = 1.00, recall = 1.00, and the per-gene allele count matched the truth for 52/52 genes.
IgDiscover, run on the raw reads with the cartridge as its starting database, independently agreed: precision = 1.00 (zero false-positive alleles), recall = 0.96 (50/52 carried alleles), with all three deletions correct and all heterozygous genes fully resolved (both alleles recovered). The two missed alleles were low-expression single-copy genes below IgDiscover's default expression threshold - a tool-tuning matter, not a simulation artefact.
These are upper-bound numbers on idealised data
Near-perfect recovery is expected here: the simulated germline names match the
scoring database exactly, SHM is light and substitution-only, and there are no
indels, chimeras, or contamination. Real data is harder - which is the point
of being able to dial difficulty up (heavier SHM via mutate, sequencing
artefacts via the corruption passes, lower per-allele depth) and re-measure
how each tool degrades against the same known truth.
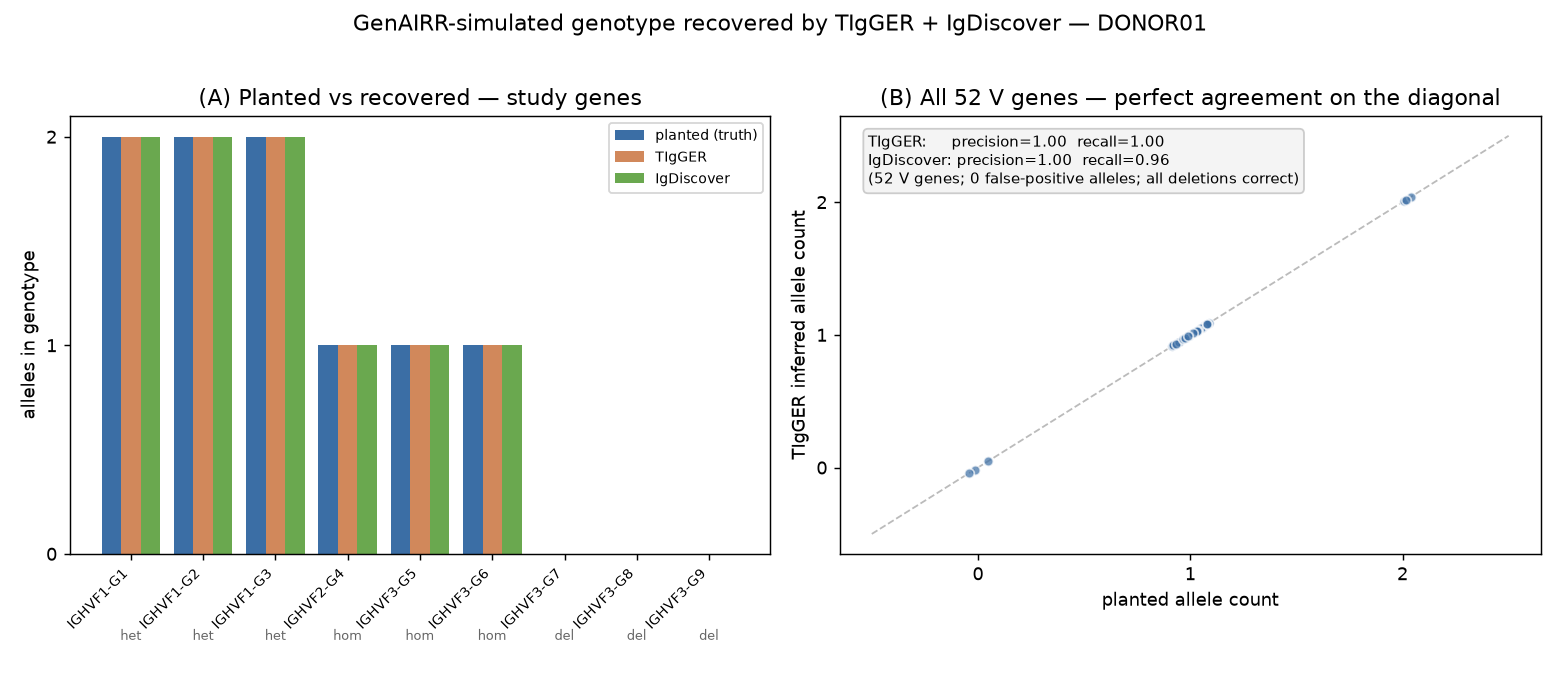
(A) The nine study genes: both tools' inferred allele counts match the planted zygosity for each (heterozygous → 2, homozygous → 1, deleted → 0). (B) All 52 V genes fall on the agreement diagonal; presence precision = 1.00 for both tools, recall 1.00 (TIgGER) / 0.96 (IgDiscover), zero false-positive alleles, all deletions correct.
Reproduce it¶
This builds the exact genotype behind the figure - 3 heterozygous, 3
homozygous, and 3 deleted study V genes, the rest filled from the reference -
simulates 4,000 reads with light SHM at seed=7, and writes every input the two
tools need plus the ground truth to score against:
import GenAIRR as ga
import GenAIRR.data as gdata
from GenAIRR.genotype import Genotype
cfg = gdata.HUMAN_IGH_OGRDB
HET = ["IGHVF1-G1", "IGHVF1-G2", "IGHVF1-G3"] # 2 alleles each
HOM = ["IGHVF2-G4", "IGHVF3-G5", "IGHVF3-G6"] # 1 allele
DEL = ["IGHVF3-G7", "IGHVF3-G8", "IGHVF3-G9"] # deleted (both chromosomes)
g = Genotype.from_dataconfig(cfg).complete_from_reference("homozygous_first_reference")
for gene in HET:
a0, a1 = (a.name for a in cfg.v_alleles[gene][:2])
g.heterozygous(gene, a0, a1)
for gene in HOM:
g.homozygous(gene, cfg.v_alleles[gene][0].name)
for gene in DEL:
g.delete_gene(gene, haplotype="both")
g.with_subject("DONOR01")
res = (
ga.Experiment.on(cfg).with_genotype(g).recombine()
.mutate(rate=0.004) # light SHM, as in real data
.run_records(n=4000, seed=7, expose_provenance=True)
)
res.to_tsv("repertoire.tsv") # AIRR table → TIgGER
g.to_tsv("truth_genotype.tsv") # ground truth to score against
with open("reads.fasta", "w") as fh: # raw reads → IgDiscover / partis
for r in res:
fh.write(f">{r['sequence_id']}\n{r['sequence'].upper()}\n")
with open("germline_V.fasta", "w") as fh: # cartridge V germline (names match v_call)
for gene, alleles in cfg.v_alleles.items():
for a in alleles:
fh.write(f">{a.name}\n{a.ungapped_seq.upper()}\n")
Score it. Run TIgGER (R snippet above) on repertoire.tsv, or IgDiscover on
reads.fasta with the cartridge as its starting database
(igdiscover init --database db/ --single-reads reads.fasta project/ && cd project
&& igdiscover run). Then compare each tool's per-gene allele set against
g.to_table() (the planted truth): allele-presence precision/recall, zygosity,
and deletion calls. With the genotype above this yields TIgGER precision/recall
1.00 (52/52 genes) and IgDiscover precision 1.00 / recall 0.96 - the figure.
Running other tools on the same data¶
The only difference between tools is whether they consume the AIRR table
(TIgGER) or the raw reads (reads.fasta, which you can write from result),
running their own aligner:
- IgDiscover - germline discovery from reads (its
own IgBLAST + iterative filtering). Initialise with the cartridge germline as
the starting database and the simulated reads, then run the pipeline; the
final/database/V.fastaexpressed-allele set is the recovered genotype:
- partis - HMM annotation with per-sample germline inference. Start from the cartridge germline and cache parameters into a parameter directory:
partis infers a per-sample germline set during this step (starting from
--initial-germline-dir) and writes it under --parameter-dir; see partis's
germline-inference docs
for the exact output location. Score that recovered allele set against
g.to_table() the same way.
Because the genotype is planted, every tool is scored identically: recovered
allele set vs genotype.to_table() - presence precision/recall, zygosity, and
deletion calls.
Where to go next¶
- Genotypes (overview) - building genotypes, novel alleles, and
the
to_table()/to_tsv()ground truth you score against. - Genotype cohorts - scale this recipe to many donors in
one
run_cohortcall.